Introduction
scAtlasVAE is a method for rapid atlas-level integration of large-scale scRNA-seq datasets and accurate data transfer for query datasets.
Installation
Install via PyPI:
1pip install scatlasvae
Install from source:
1 git clone git@github.com:WanluLiuLab/scAtlasVAE.git
2 cd scAtlasVAE
3 pip3 install -r requirements.txt
4 python3 setup.py install
PyTorch installation
If you have a different CUDA version, you can check for the PyTorch version that is compatible with your CUDA version [here](https://pytorch.org/get-started/previous-versions/).
scAtlasVAE has not been tested with other backend including mps.
Tutorial
Basic Usage
Integrating scRNA-seq data using scatlasvae.model.scAtlasVAE is straight forward and compatible with scanpy’s anndata.
1import scatlasvae
2
3# Load the data
4adata = scatlasvae.read_h5ad("path/to/data.h5ad")
5vae_model = scatlasvae.model.scAtlasVAE(
6 adata=adata,
7 batch_key="sample_name",
8)
9vae_model.fit()
See Integrating Multi-source Gene Expression (GEX) for more details. See Transfering Multi-source Gene Expression (GEX) for more details.
Key functionality

scAtlasVAE can be used to build reference atlas without prior cell type annotation. The reference atlas can be used for downstream tasks such as query-to-reference mapping and multi-atlas integration. See Integrating Multi-source Gene Expression (GEX) for more details.`

scAtlasVAE can be used to map query dataset to reference atlas with cell type annotation. The query dataset can be used for downstream tasks such as multi-atlas integration. See Transfering Multi-source Gene Expression (GEX) for more details.

scAtlasVAE can be used to build reference atlas without prior cell type annotation. The reference atlas can be used for downstream tasks such as query-to-reference mapping and multi-atlas integration. See Integrating Multi-source Gene Expression (GEX) for more details. See Tools for more details.
Model architecture
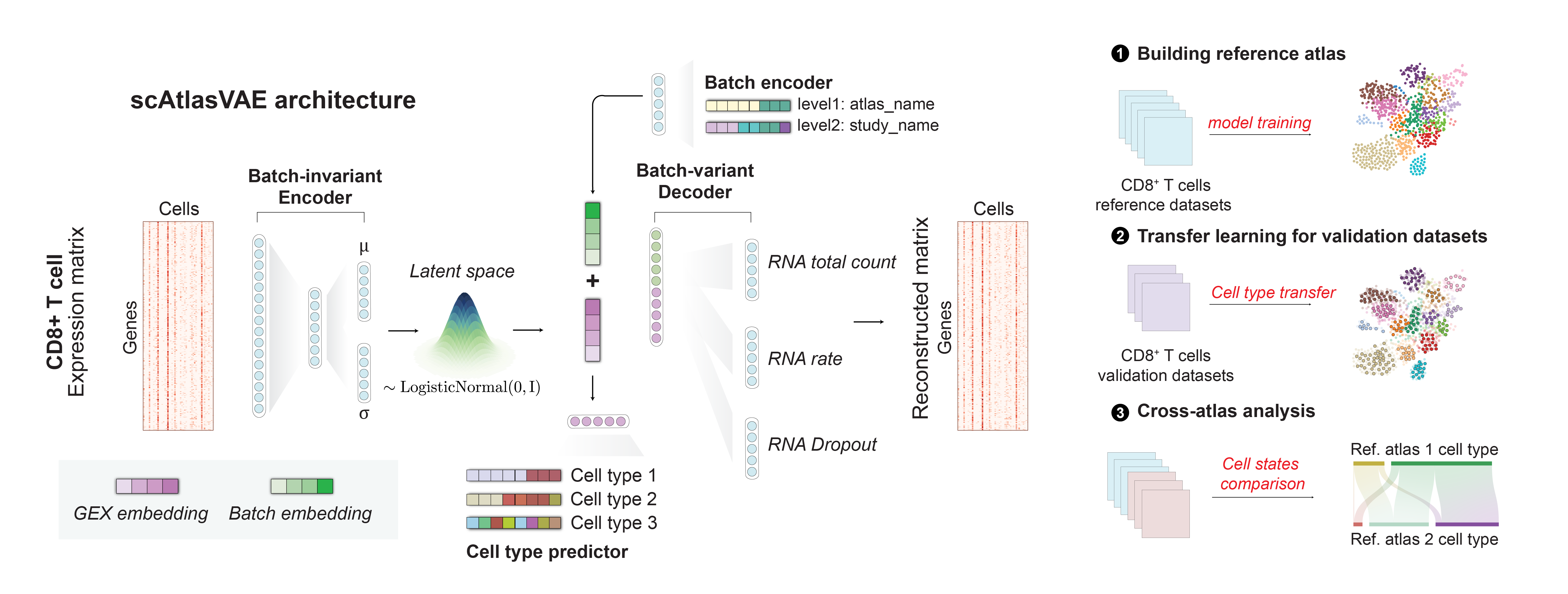
The scAtlasVAE model is based on a variational autoencoder with a batch-invariant encoder and a batch-variant decoder. The batch-invariant encoder accepts raw gene expression count transformed by NormalizeTotal operation (optional) and Log1p operation as input. The multi-layer perceptron (MLP) of the encoder project the gene expression matrix to an mean and variance of the latent representation of each cell. the batch index of each cell is annotated as \(\mathbf{B}\ =\{ {B}_1,\ldots,B_n \} \in \mathbb{R}^H,\ B_n=\ \{ b_{n,1},\ldots,b_{n,L} \}\), where \(L\) is the number of levels of batches. If cell subtype annotation is available, the label index of as \({\mathbf{C}\ = \{C}_1,\ldots,C_n \}\in\mathbb{R}^A,\ C_n\ =\{C_{n,1},\ldots,C_{n,A}\}\) where \(A\) is the number of categories of annotations.
The batch-variant decoder is composed of an embedding module that embeds the batch information \(\mathbf{B}\) to an batch embedding and a MLP layer that accepts the concatenated embedding of the cell latent representation \(\mathbf{z}\) and the batch embedding. The MLP layer outputs the mean, variance, and dropout probability (\(r_{mean},r_{var},r_{gate}\)) of the gene expression value for each cell for fitting an zero-inflated negative binomial (ZINB) distribution.
scAtlasVAE was trained on random sampled batches of datasets to minimize the log-likelihood on the observed single-cell sequencing data \(\mathbf{X}\) and by default, we use zero-inflated negative binomial distribution:
scAtlasVAE includes cell type predictor \(\mathcal{F}_{\mathrm{celltype}}(\mathbf{z})\ = \{{f_{\mathrm{celltype}}}_1,\ldots,{f_{\mathrm{celltype}}}_A \}\) for independent prediction of different cell type categories. The cell type loss is defined as
where \(C_a\) is the number of cell types in category a, \({f_{{celltype}_a}\left(x_{a,n}\right)}_c\) is the logits of cell type prediction for cell type \(c\), \({\hat{y}}_{a,n}\) is the ground truth cell type of category \(a\) for cell \(n\). \({w_{\hat{y}}}_{a,n}\) is the weight of cell type \({\hat{y}}_{a,n}\) inversely proportional to the number of cell types in category \(a\). The \({ \mathrm{ignore\_index}}_a\) indicates whether the cell type a is available for cell \(n\).